
CutProtFam-Pred: an on-line tool for detection and classification of putative structural cuticular proteins from sequence alone, based on profile Hidden Markov Models
Ioannidou Z.S., Theodoropoulou M.C., Papandreou N.C., Willis J.H. and Hamodrakas S.J. (2014). "CutProtFam-Pred: detection and classification of putative structural cuticular proteins from sequence alone, based on profile hidden Markov models." Insect Biochem Mol Biol 52: 51-59. [PudMedID: 24978609]
AND
Karouzou M.V., Spyropoulos Y., Iconomidou V.A., Cornman R.S., Hamodrakas S.J. and Willis J.H. (2007). "Drosophila cuticular proteins with the R&R Consensus: annotation and classification with a new tool for discriminating RR-1 and RR-2 sequences." Insect Biochem Mol Biol 37(8): 754-760. [PudMedID: 17628275]
The arthropod cuticle, which acts as an exoskeleton or contributes to tracheae and linings of the fore- and hind-gut and some other structures, is a composite, bipartite system, made of chitin (polysaccharide of N-acetylglucosamine) filaments embedded in a proteinaceous matrix. The physical properties of cuticle, such as its complex structure, both strong and flexible, are determined by the structure of its two major components, cuticular proteins (CPs) and chitin, and, also, by their interactions. The architecture of the cuticle is helicoidal and most probably it is responsible for cuticle's extraordinary mechanical and physiological properties. In this helicoidal structure, chitin is in the form of crystalline filaments and proteins play the role of the matrix. (Neville, 1975)
The proteinaceous matrix consists mainly of structural cuticular proteins, which differ between cuticles of different types. Their quantitative distribution may change, and many can be classified into one of 13 protein families, based on some conserved amino acid regions. The majority of the structural proteins that have been discovered to date belong to the CPR family, and they are identified by the conserved R&R region (Rebers and Riddiford Consensus), which is recognized by PF00379, the Pfam motif for chitin binding of arthropod cuticle and it is most probably dominated by β-sheet structure. Three sub-families of the CPR family RR-1, RR-2 and RR-3, have also been identified from conservation at sequence level and limited correlation with the cuticle type. RR-1 is found in proteins from soft cuticles, and RR-2 in proteins from hard cuticles, while RR-3 has been found in very few sequences. Also, the secondary structure prediction for the RR-1 and RR-2 types, verifies the fact that they appear in region of cuticle with different properties.
Recently, several novel families, also containing characteristic conserved regions, have been described: CPF (based on a conserved region with 44 amino acids); CPFL (with a conserved C-terminal region similar to CPF); the low complexity families: Tweedle, CPLCA, CPLCG, CPLCP, CPLCW; CPCFC (2 or 3 C-x(5)-C repeats); CPG (rich in glycines); CPAP3 and CPAP1 (analogous to peritrophins, with 3 and 1 chitin-binding domains respectively). Some of these families are restricted to diptera or even mosquitoes. Willis et al. (2012) and Willis (2010) offer insights for all thirteen families and describe extensively each family's features, in detailed reviews.
The package HMMER v3.0 (Eddy, 1998) (http://hmmer.janelia.org/) was used to build characteristic profile Hidden Markov Models based on multiple alignments of these conserved regions, for CPF, CPCFC, CPLCA, CPLCG, CPLCW, Tweedle, CPAP3 and CPAP1 families. There was insufficient sequence conservation to model the other families. Also, some proteins isolated from cuticle have not been assigned to families, but both the families without models and sequences not assigned to familes, at present, are a minority of the structural cuticular protein sequences.
Apart from the PF00379 Pfam motif, there was already available a tool for distinction between the RR-1 and RR-2 types of CPR, which as referred above, constitute the majority of the CPR proteins.
The aim was to make an on-line tool that could be applied to sequence alone that allows the accurate detection of structural cuticular proteins and specifically distinguishes among the new families, in addition to the old ones.
It is hoped that this tool will be of help to proteome annotators as more arthropod proteomes become available.
For several reasons, the data produced using the tool must be considered as only a preliminary estimate and aid to annotation and CP identification in proteomes, not as a substitute for manual annotation.
First, there is the problem that not all recognized families yielded sequences that could be used to develop tools.
Then there are authentic cuticular protein sequences that have not been assigned to families.
Finally, even for families the tool recognizes, there is the limitation that the tool queries proteomes that have been produced by automated annotation. These programs sometimes combine closely linked genes into a single protein and many cuticular proteins are tightly clustered on chromosomes. HMMER produces scores based on the number of hits as well as their quality. Since only rarely does a CPR protein have more than a single R&R Consensus region, high ranking proteins are apt to have been incorrectly annotated due to combining adjacent genes. Thus, even with the recommended low setting for CPRs that will recognize RR-3 sequences, proteins combined by incorrect annotation mean that the total number of identified CPRs is almost certain to be an underestimate. Also, with this low setting, both RR-1 and RR-2 proteins will be identified, so duplicates must be eliminated and the RR type with the best score selected. If you search for all families, the low score is used and then the best type automatically selected.
****NEW (Added on May 2023): When proteins have been assigned to either the CPAP1 or CPAP3 families by our tool, it is essential that the sequence be checked to be sure they have no more than 1 or 3 CBM_14 domains, respectively. This can be done quickly by using NCBI Conserved Domain Search [https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi].
****NEW (Added on May 2023): When proteins have been assigned to either the CPAP1 or CPAP3 families by our tool, it is essential that the sequence be checked to be sure they have no more than 1 or 3 CBM_14 domains, respectively. This can be done quickly by using NCBI Conserved Domain Search [https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi].
In order to use the tool, the user should press the "SEARCH" tab. A form appears with multiple options.
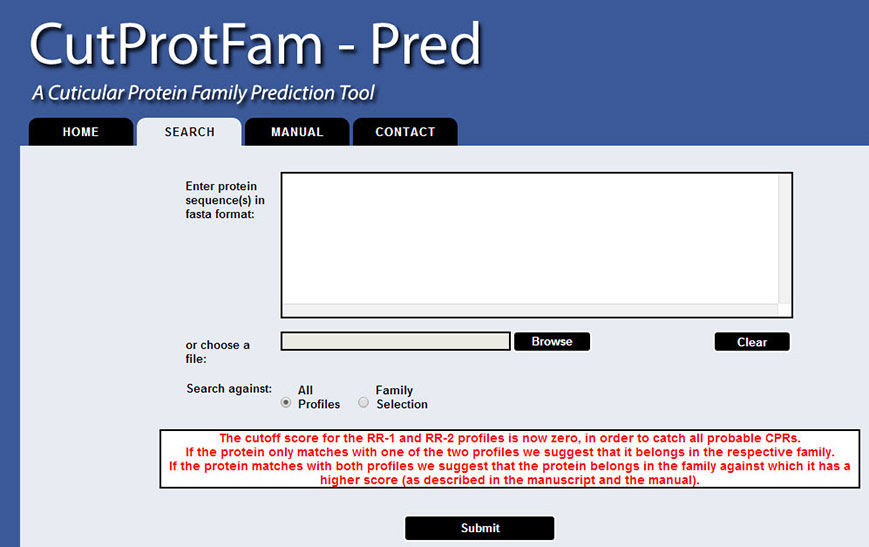
The base of the CutProtFam-Pred predictor is a library of profile Hidden Markov Models (pHMMs), one for each family, that have been trained with/by multiple sequence alignment of the conserved region of the corresponding structural cuticular protein family.
Of the 12 families other than CPR, the construction of characteristic profiles was possible for 8 of them using the HMMER software package version 3.0 (Eddy, 1998).
For the 13th family (CPR), the profiles that were already available in cuticleDB (Magrioti et al., 2004) for RR-1 and RR-2 (Karouzou et al., 2007) were used; those had been created using the HMMER software package version 2.3.2 (Eddy, 1998).
The user may submit a list of fasta-formatted protein sequences and search if they fit one of the available profiles that describe the structural cuticular protein families.
Generally, fasta format is a text-based format for representing peptide (or nucleotide) sequences, in which amino acids (or nucleotides) are represented using single-letter codes, and it also allows for sequence names and comments to precede the sequences.
A sequence in fasta format begins with a single-line description, followed by lines of sequences data. The description line is distinguished from the sequence data by a greater-than (">") symbol in the first column. The word following the ">" symbol is the identifier of the sequence, and the rest of the line is the description. There should be no space between the ">" and the first letter of the identifier. The sequence ends if another line starting with a ">" appears; this indicates the start of another sequence.
(http://zhanglab.ccmb.med.umich.edu/FASTA/)
For this tool, the identifier is mandatory and must be unique. Lower-case letters in the sequence are accepted and are mapped into upper-case. Only the amino acid symbols are allowed (ABCDEFGHIKLMNPQRSTUVWYZ), plus the X character that stands for unknown amino acid (*s and -s are now allowed too).
The sequence data can be submitted in two ways. The user can either copy and paste a fasta-formatted sequences in the textbox area, or upload a file containing fasta-formatted sequences.
The selections can be cleared in case of a mistake using the "Clear" button.
If both options are filled, the tool will use the uploaded file only, ignoring whatever is written inside the textbox.
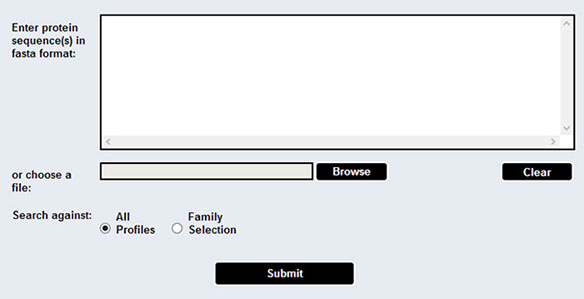
The user can submit up to x line of sequences using the text box, or upload a file containing sequences up to x MB. To perform a large scale search using a larger file or a whole proteome, please contact us! (See CONTACT tab.) All submitted data are kept confidential and they are deleted upon one week after submission.
The user can search sequences for all families against a library of profiles, using each family's default cutoff.
Our evaluation of each model revealed it to be specific for the corresponding family or for the subfamily in the case of RR-1 and RR-2.
In case more than one model fits a sequence, the one with the higher score will be chosen, unless one of the matches is to the CPR family for then all other matches will be ignored.
[HMMER's hmmpfam (version 2.3.2 for CPR_RR-1 and CPR_RR-2) and hmmscan (version 3.0 for all the other families) are used.]
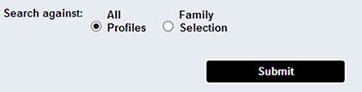
The user has the option to choose to search sequences against a specific family profile, and use either the default cutoff score (which appears as the default value in the box), or input a different, user selected, cutoff value, in the form of score or e-value.
[HMMER's hmmsearch (version 2.3.2 for CPR_RR-1 and CPR_RR-2, and version 3.0 for all the other families) is used.]
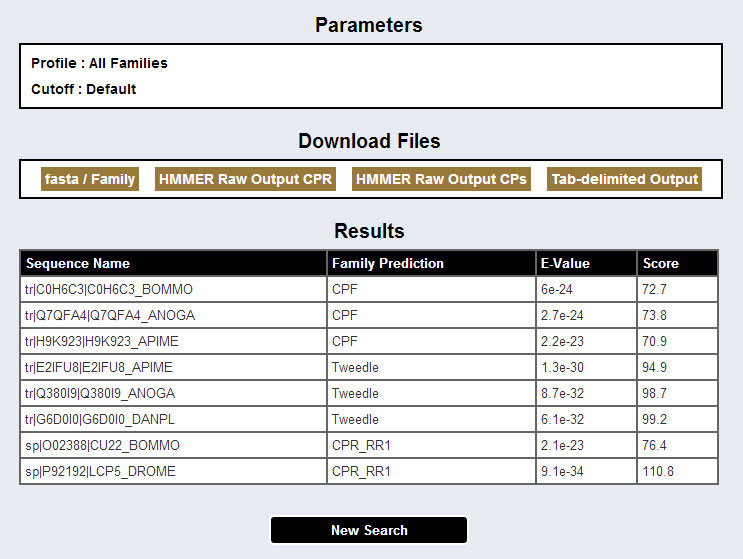
In the results page, there is table containing following columns: the first column is the name of the sequence, the second column is the prediction (type) for the sequence, and the third and the fourth columns are the e-value and the score of the prediction.
The score is the log-odds score for the complete sequence; a positive score shows a positive correlation.
The e-value is the statistical significance of the match to this sequence, the number of hits we'd expect to score this highly in a database of this size if the database contained only non-homologous random sequences; the lower the e-value, the more significant the hit.
The scores and the e-values used as representing for the predictions are the "full sequence" ones in all cases except the CPAP1 and CPAP3 families where the "best domain" scores and e-values are used instead (for more details see Ioannidou et al., 2014).
The user can copy and paste this table directly into a spreadsheet program (for example Microsoft Excel, OpenOffice Calc, Numbers for Mac, etc.), download it in a tab-delimited format (which can be opened with either a spreadsheet program or a text editor, or even be parsed using a programming language), and/or download the raw HMMER output, and/or download the protein sequences, for those sequences where there was a prediction, in fasta format files, separated by protein family.
****NEW (Added on May 2023): When proteins have been assigned to either the CPAP1 or CPAP3 families by our tool, it is essential that the sequence be checked to be sure they have no more than 1 or 3 CBM_14 domains, respectively. This can be done quickly by using NCBI Conserved Domain Search [https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi].
****NEW (Added on May 2023): When proteins have been assigned to either the CPAP1 or CPAP3 families by our tool, it is essential that the sequence be checked to be sure they have no more than 1 or 3 CBM_14 domains, respectively. This can be done quickly by using NCBI Conserved Domain Search [https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi].
Eddy, S.R. (1998). "Profile hidden Markov models." Bioinformatics (Oxford) 14(9): 755-763. [PudMedID: 9918945]
Ioannidou Z.S., Theodoropoulou M.C., Papandreou N.C., Willis J.H. and Hamodrakas S.J. (2014). "CutProtFam-Pred: detection and classification of putative structural cuticular proteins from sequence alone, based on profile hidden Markov models." Insect Biochemistry and Molecular Biology 52: 51-59. [PudMedID: 24978609]
Karouzou M.V., Spyropoulos Y., Iconomidou V.A., Cornman R.S., Hamodrakas S.J. and Willis J.H. (2007). "Drosophila cuticular proteins with the R&R Consensus: annotation and classification with a new tool for discriminating RR-1 and RR-2 sequences." Insect Biochemistry and Molecular Biology 37(8): 754-760. [PudMedID: 17628275]
Magkrioti, C.K., Spyropoulos, I.C., Iconomidou, V.A., Willis, J.H., and Hamodrakas, S.J. (2004). "cuticleDB: a relational database of Arthropod cuticular proteins." BMC Bioinformatics 5: 138. [PudMedID: 15453918]
Neville, A.C. (1975). Biology of the arthropod cuticle. Berlin ; New York, Springer-Verlag. [Link]
Willis, J.H. (2010). "Structural cuticular proteins from arthropods: annotation, nomenclature, and sequence characteristics in the genomics era." Insect Biochemistry and Molecular Biology 40(3): 189-204. [PudMedID: 20171281]
Willis, J.H., Papandreou, N.C., Iconomidou, V.A., and Hamodrakas, S.J. (2012). "5 - Cuticular Proteins". Insect Molecular Biology and Biochemistry. I. G. Lawrence. San Diego, Academic Press: 134-166. [Link]